摘 要
鈦鋁介金屬合金因質輕且具高溫強度, 是極具潛力的航空用高溫材料。但在高溫結構應用上, 除須考慮高溫機械性質外, 材料之化學特性, 尤其是高溫氧化與腐蝕亦是必須了解的問題。
本研究是針對Ti3A1-Nb合金討論其在600 ~ 900℃, 空氣中的高溫氧化行為。表面氧化層結構藉X光繞射, SEM電子顯微鏡及EPMA電子顯微探測進行分析和觀察。
研究結果顯示, Ti3A1合金在700℃因氧化層剝落已喪失其抗氧化性。添加Nb的合金由於氧化物晶粒細化, 形成較緻密的氧化層; 並加速鋁原子向外擴散, 增加表面的保護性及抗氧化能力, 所以Nb含量愈高的鈦鋁合金, 其抗氧化性愈佳。
Ti3A1-Nb合金添加微量的Si和Cr可以降低氧化速率。而含Y的Ti3A1-Nb合金必須到900℃才有助於抗氧化能力, Y可使表面氧化層的附著力增強, 不易剝落。
Ti3A1-Nb合金的氧化行為是以拋物線的氧化速率進行, 可知其氧化控制機構為離子在氧化層裡的擴散。
關鍵詞: 鈦鋁介金屬合金, 高溫氧化行為, 抗氧化能力
1. 前言
鈦鋁介金屬合金的發展已有長遠的歷史。由於它具有優良的高溫特性, 如抗潛變、耐高溫疲勞等性質非常優異, 再加上質量輕, 因而成為極具潛力的高溫材料, 在航太材料的應用和設計上, 提供了另一較佳的選擇。
從1985年Lipsitt[1]等人的研究迄今, 許多學者的研究主要是在探討鈦鋁介金屬合金的低溫脆性。由於介金屬特殊的序化結構, 使之有優秀的高溫機械性質; 但在常溫時, 卻因差排活性小及其有限的滑移系統, 使鈦鋁合金( Ti3A1)幾乎沒有發生應變而脆斷[2], 因此無法以普通的製程方式來加工, 這是一大缺憾。
考慮鈦鋁介金屬合金的高溫應用時的另一個重要問題即是高溫氧化。處在高溫狀態時, 避免不了氧化。Lipsitt[1]曾報導Ti3A1合金的抗氧化溫度約為650℃; 即表示此一材料在高於650℃時, 失去強度前即已無法抵抗高溫氧化。若增加合金的鋁含量, 雖然可以增加表面氧化鋁的含量(成份必須在63ᵂ/ₒ Al以上, 才能形成連續的A12O3層[3][4]), 但卻會使鈦鋁介金屬合金的脆性問題更形嚴重。最近, 日本人針對1:1的鈦鋁介金屬合金做了頗多的努力, 包括表面處理[5](Coating), 在低氧分壓下熱處理[6]及添加Si, Mn, Cr和Y等第三元合金元素[7]來改善鈦鋁合金的高溫氧化問題。
Ti3A1基介金屬合金的研究存有常溫脆性和高溫氧化的兩大問題。目前的研究大部分係以添加鈮(Nb)的鈦鋁介金屬合金作為主題。而添加鈮的合金效應, 在機械性質方面, 可使差排活性增加, 改善常溫的延性[8][9]。在一系列Ti3A1-Nb合金中, 已發現Ti65Al25b10成份的合金有較突出的延性, 特別是經過熱軋處理後延性更佳[10]。另外, Ti3A1-Nb合金的顯微組織也因鈮的關係變得較複雜; 除了主要的α₂(Ti3A1,DO19)和β(B2結構)兩相組成外, 還有o相[11](orthorhombic system, Cmcm)與准穩定(metastable)的ω相[12](B82結構)。這些相變化和熱處理過程之間的關係可藉Banerjee[13][14]等人提出的局部Ti3A1-Nb相圖與TTT曲線來說明。(如圖1/2/3所示)
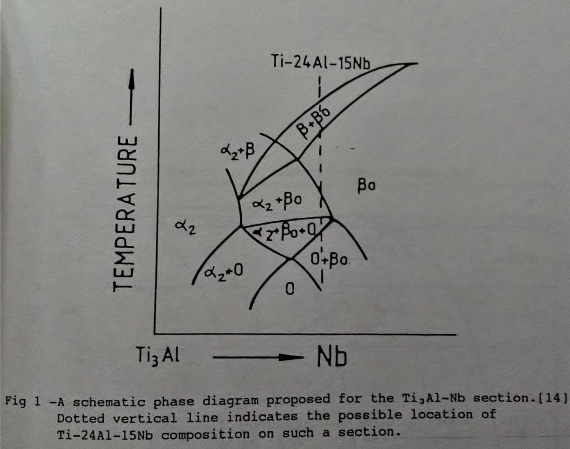
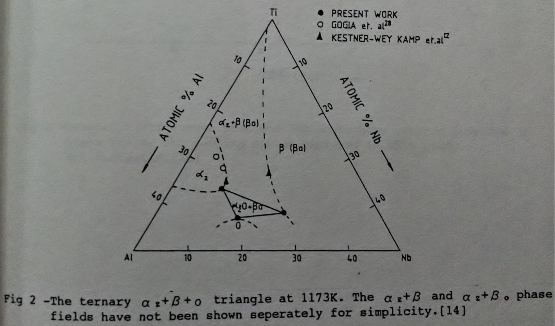
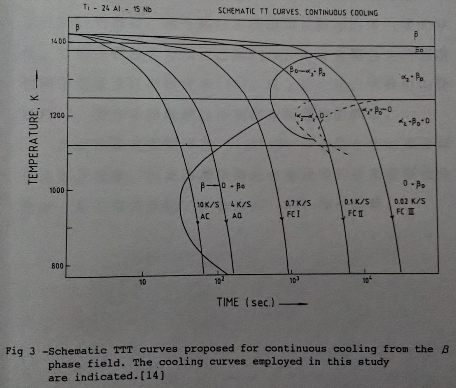
有關Ti3A1-Nb介金屬合金的高溫氧化研究, 目前只有少數的幾篇論文[15][16]可供參考。他們的研究結果顯示鈮可以提高鈦鋁介金屬合金的抗氧化能力, 但其高溫氧化行為或機構尚不十分了解。本文的目的即在探討Ti3A1-Nb合金的高溫氧化行為,觀察表面氧化層的分布, 並試著添加微量的第四元合金元素以增進Ti3A1-Nb合金的抗氧化能力等。Ti3A1基合金的應用性, 已經過美國空軍的評估[17], 包括精密鑄造, 壓延和鍛造, 及超塑性成形, 擴散接合等關鍵的技術。鑄造方面, 報導指出已經成功地生產了815Kg的鑄錠, 而且藉精密鑄造可以製造許多引擎的零件。目前我們臺大方面也已對Ti3A1-Nb介金屬合金的超塑性與擴散接合技術研究中[18]。以上的介紹,大致可勾勒出Ti3A1基合金的發展沿革和現在所遭遇的問題。
本文的研究重點將在Ti3A1-Nb介金屬合金的高溫氧化速率與機構, 以及添加第四元合金元素的抗高溫氧化效應等, 最後再與鈦鋁二元合金( Ti3A1)作一比較。
2. 理論探討
1. 高溫氧化機構[20]
目前研究Ti3A1-Nb介金屬合金的氧化動力學, 大都沿用拋物線型的氧化速率模式, 亦即Wagner提出的離子或點缺陷在氧化層裡擴散所控制的氧化機構理論[21]。
若考慮金屬的氧化反應: M(s)+½O2(g)=MO(s)
反應的產物MO會在金屬表面生成, 為了使反應繼續進行, 反應物就必須穿越氧化層才能在金屬和氧化物的界面或者氧化物與氣體的界面上發生反應。因此反應物經由氧化物的質量傳送, 亦即擴散的過程, 就變成氧化時的一個重要步驟。
由於擴散的粒子包含陰離子、陽離子及點缺陷, 而在氧化層中質傳方式決定於何種粒子主導? 歸納出可能以下列的兩種方式進行(如圖4)。另外要維持電荷數目一定, 必須假設氧化物為非化合比的化合物(Non-stochiometric compound), 例如M₁±δO, δ為一極小值。以下即分別討論兩種粒子擴散的方式:

A. N型氧化物
此種氧化物之化學式為M₁₊δO, 表示金屬陽離子或氧空位(oxygen vacancy)過多, 由它們來主導氧化時的擴散。舉氧化鋅, ZnO, 為例, 在晶格間隙的鋅離子會向外移動, 進行氧化反應:
Znᵢ⁺⁺ + 2e⁻ + ½O₂ = ZnO ...........(1)
Znᵢ⁺⁺: 格隙中的鋅離子, e⁻: 電子
以圖5表示其過程。
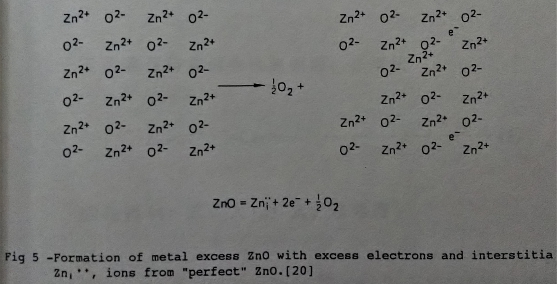
第(1)式的平衡常數, K, 可表示為:
K=aZnᵢ⁺⁺∙ae⁻²∙PO₂½ ..........(2)
假設Znᵢ⁺⁺數目很少, 視為一稀薄溶液, 遵守亨利定律, (2)式可改寫成:
K=CZnᵢ⁺⁺∙Ce⁻²∙PO₂½ ..........(3)
Ci: i粒子的濃度
且因為保持電價中性的原則, 必須使
2CZnᵢ⁺⁺=Ce⁻ ..........(4)
因此將(4)式代入(3)式, 整理得
K=4CZnᵢ⁺⁺³∙PO₂½ or Cznᵢ⁺⁺=(K/4)⅓∙PO₂⁻⅙
⸫Cznᵢ⁺⁺=½Ce⁻ ~ PO₂⁻⅙ ..........(5)
所以藉量測氧化層的電阻值或氧分壓的關係, 即知道是幾價的離子(Znᵢ⁺ or Znᵢ⁺⁺)在氧化層中擴散。
另外在N型氧化物中, 也會產生氧原子晶格上的空位, VO²⁻, 不過, 其擴散方向與金屬陽離子相反。
B. P型氧化物
P型氧化物, M₁₋δO, 由於缺少金屬陽離子易形成陽離子空位, VM²⁺, 同時陽離子的外層電子軌域接近, 易被激發而改變價數, 譬如Ni²⁺↔Ni³⁺即是。因此舉氧化鎳NiO為例, 其擴散和氧化過程如圖6所示。氧化層中形成鎳晶格空位, VNi²⁻, 同時有些鎳離子由二價變成三價(Ni³⁺)鎳離子, 可以提供電子的低能階位置, 因此被稱為⸢電洞⸥(electron hole), 使鎳離子往外移動, 而VNi²⁻則向反方向移動。以上的過程可以下列的反應式表示:

½O₂ = OO + 2h⁺ + VNi²⁻ ..........(6)
OO: 位於氧化物晶格上的氧原子
h⁺: 電洞, 亦是Ni³⁺的位置
VNi²⁻: 帶二價負電的 鎳離子空位
關於 P型氧化層之電阻值與氧分壓的關係如前述推導, 結果為:
Ch⁺ = const∙ PO₂⅙ ..........(7)
2. 氧化速率
描述氧化動力學的方程式, 理論上有三種型態[21]:
n=1, 直線型(linear)
Wn=k∙t, n=2, 拋物線型(parabolic) ..........(8)
n=3, 立方線型(cubic)
W: 單位面積的重量變化
n: 氧化指數
k: 速率常數
t: 時間
高溫氧化機構多是先以擴散控制的拋物線型氧化動力學模式來探討, 所以焦點將集中在W²=kₚ∙t氧化速率方程式。
通常高溫氧化的進行是時間和溫度的函數, W²=kₚ∙t式中, 速率常數kₚ應為溫度的函數, 它與擴散係數相似, 適用於Arrhenius關係式:
kₚ=k0∙exp(-Q/RT) ..........(9)
k0: 常數, Q: 氧化所需的活化能, T: 溫度/K, R=8.3143 J/mole∙K
將(8), (9)兩式結合,可改寫成:
W²=k0∙t∙exp(-Q/RT) ..........(10)
3. Ti-A1介金屬合金的高溫氧化
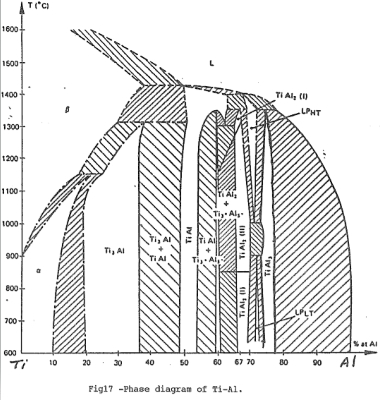
- 鈦鋁金屬高溫氧化時, 鋁的多寡會影響其氧化速率的快慢。一般而言, 抗氧化能力隨鋁含量增加而加強, 研究[3][4]指出鋁增加至63ᵂ/ₒ 以上即可形成連續的氧化鋁保護層。另外就Ti-A1隨成份辦化的活性趨勢來看[22](如圖7), 在Ti3A1和TiA1的區域, 鋁的活性偏低所以表面形成TiO₂氧化層。但在TiA13(約63ᵂ/ₒA1)的區域, 鋁的活性變大, 因此形成A12O3保護層。
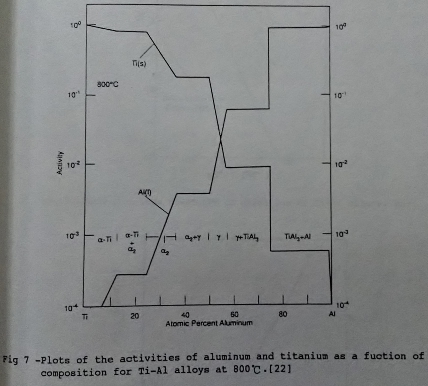
從Arrhenius圖(logkₚ vs. 1/T plot)去分析, 如圖8[23], 兩側的直線分別為TiO₂和A12O3的Arrhenius關係, 隨鋁的增加, 鈦鋁合金的線性向形成A12O3的直線靠近。
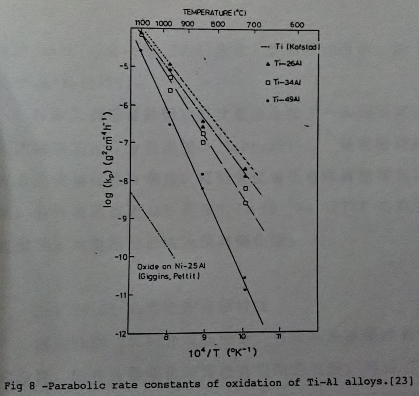
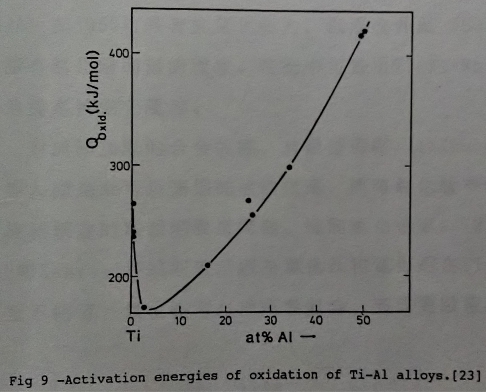
圖9所示的是氧化所需的活化能[23], Qoxid., 隨鋁增加而變大, 這是鋁含量增加有助於抗氧化性的另一項佐證。
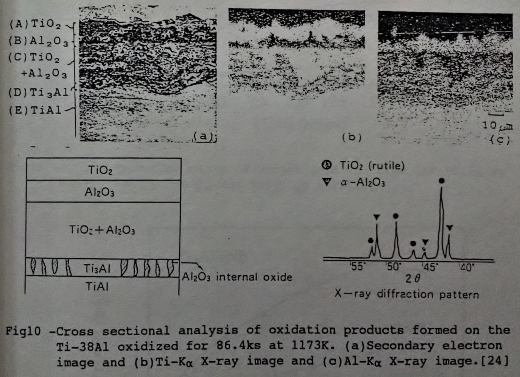
氧化層的結構, 在富鈦的Ti-A1合金來說, 最外層大部分是氧化鈦, 往內是TiO₂+A12O3或A12O3的組織, 接近基地(matrix)有一層Ti3A1分布, 詳如圖10所示, 以Ti-38ᵂ/ₒA1為例[24]。
日本人最近曾針對1:1Ti-A1合金做了一系列研究。譬如添加第三元合金元素如Cr, Y, Mn, Si[7], 發現矽對鈦鋁合金的抗氧化性有幫助, 釔則有使氧化層附著性增加的效果。另外表面處理[5]與在6.7×10⁻³Pa, 1273K的真空熱處理[6]均有助於形成連續的氧化鋁。
4. Ti3A1-Nb合金的化學特性
添加Nb的Ti3A1介金屬合金, 可使其常溫脆性有明顯的改善, 同時高溫氧化性質也有相同的效果。可將Ti3A1和Ti3A1-Nb兩種合金作一比較, 如圖11/12所示[25]。Ti3A1在650℃尚有抗氧化能力, 但溫度升至750℃以上, 即有氧化層剝落的現象。在圖中可看出Ti3A1-Nb合金明顯地提高抗氧化溫度。
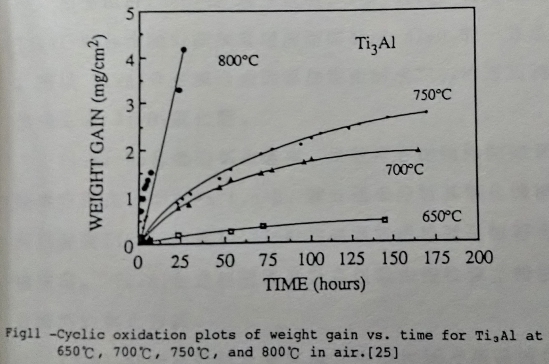
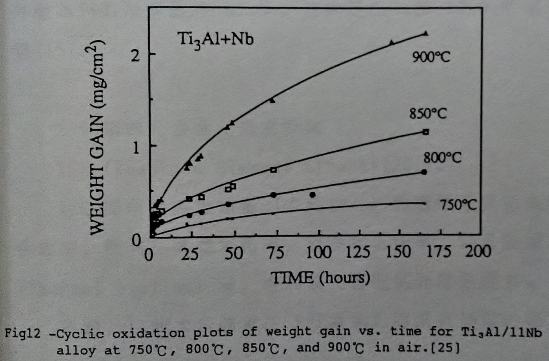
對於添加Nb的合金效應, 有幾種解釋: (1)Khobaib[15]等人認為Nb可以讓鋁的活性提高, 同時氧化層中的鈮會使控制擴散的點缺陷濃度降低, 抑制氧化速率。(2)Welsch和Kahveci[23]則表示因為氧化鈦和氧化鋁在1140℃以下互不相溶, 氧化鈮可以使兩者混合, 形成更緻密的氧化層, 而且Nb₂O₅會降低氧在氧化層的擴散速度。
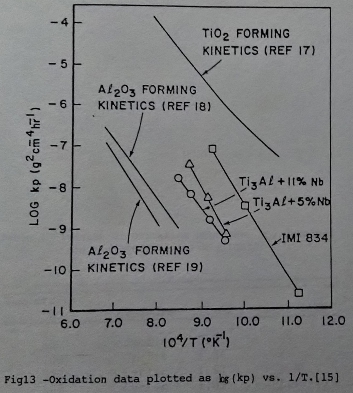
另外由Arrhenius圖來說明(圖13/14)[25][15], Ti3A1-Nb合金隨鈮添加量增加而向形成A12O3的一方逼近, 所以Ti3A1介金屬合金因添加鈮遊形成TiO₂的方式轉而傾向生成A12O3氧化層。
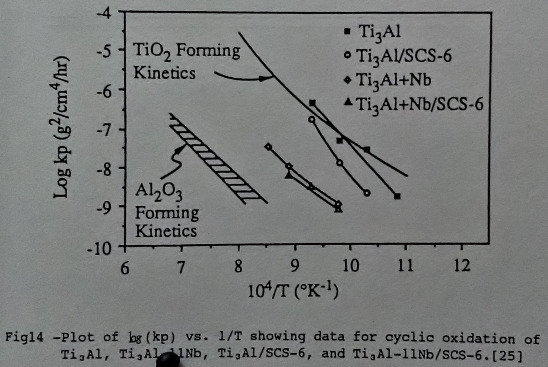
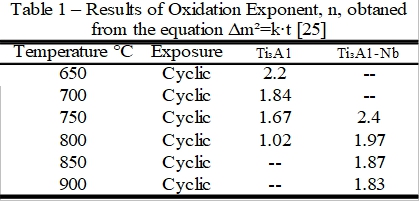
Ti3A1-Nb合金的氧化速率, 是較符合拋物線型的氧化速率方程式, 即Wn=k∙t, n=2。就n值來分析其氧化機構, 可以從表1[25]知道Ti3A1-Nb的高溫氧化確實是較符合拋物線型。Ti3A1合金則隨溫度升高逐漸由拋物線型轉換成直線型的氧化方式。
Ti3A1-Nb合金的氧化層結構, 主要包括三個區域: 最外層為TiO₂及少數的A12O3, 往內是TiO₂和A12O3的混合層。
5. 第四元合金元素之添加
REE(Reactive Element Effect)[26][27]
REE即所謂添加微量(少於1ᵂ/ₒ)的活性元素於高溫合金中, 使其在高溫氧化時, 減緩氧化速率, 以及熱循環(thermal cycle)時, 增加氧化層抵抗剝落的能力。
不過, 目前大多偏重在REE對氧化層抵抗剝落的影響。而提出的解釋則是因為活性元素或其氧化物能夠提供氧化物成核的地方(nucleation site)和成長的機會, 亦即Pettit[28]所謂的釘掛效應(Keying oe Pegging effect)。或者活性元素可以吸收空位(vacancy sink), 避免形成金屬與氧化物之間的空洞, 有損其附著性[29]。為了增加氧化層的附著力, 我們選擇釔作為添加的元素。
另外亦選擇Si和Cr作為添加的合金元素。日本的研究[7]證實Si及Cr對鈦鋁合金的抗氧化性有益。本文中我們嘗試分別添加微量的Si, Cr, Y至Ti3A1-Nb介金屬合金, 以評估第四元合金元素對高溫氧化的影響。
3. 實驗方法
1. 合金的配製與熔煉
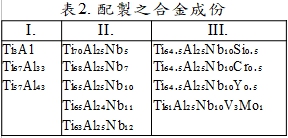
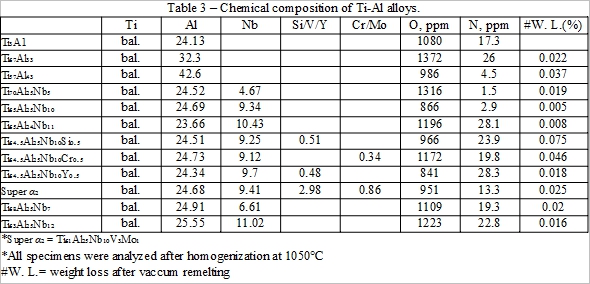
合金配製是以Ti₆₅Al₂₅Nb₁₀的成份為基準, 或添加微量的第四元合金元素, 或改變Ti-Al-Nb三元合金中的鈮含量, 並與Ti-Al合金相比較。上述合金成份如下表所列。
所選用的生材包括鈦棒(純度≈99.6%, 含氧量約1700ppm), 鋁板(純度99.9%), 鈮丸(純度約99.9%), 及其它純合金元素。
鈦和鋁在配製前先經過機械研磨去掉氧化部分, 再分別酸洗及鹼洗, 最後用蒸餾水清洗, 再烘乾即可。詳細步驟可參考[29]。
熔煉之前應將生材按照合金成份準備好, 本實驗室以非消耗性鎢極之真空電弧熔解爐來配製合金,如圖15所示。

在熔煉合金時, 須注意的是鈦, 鈮與鋁的熔點差距大, 而且鋁蒸氣壓較高, 故意揮發。因此熔煉初期可以先使鈦、鈮互熔, 再將鋁塊加入, 利用熔融金屬液使鋁熔化, 避免電弧直接打擊鋁塊使鋁揮發, 而造成合金成份偏離過大。在熔煉後, 計算重量損失來決定下回熔煉的添加量。
2. 合金的金相組織熱處理
熔煉後之試片用砂紙研磨至2000號。浸蝕以後以光學顯微鏡觀察顯微組織, 並用Vickers硬度機以300g荷重做硬度測試, 取二十點之平均。
為了消除鑄造凝固時產生之偏析效應, 試片做一1050℃, 10⁻⁵Torr的均質化熱處理。處理後之試片同樣以光學顯微鏡觀察並測試硬度值。並將試片做EPMA定量分析及氧含量測試。
3. 高溫氧化速率量測
高溫氧化的試片經過均質化熱處理之後, 切割成約10×12×1mm的小試片。氧化測試前, 試片先研磨至1000號砂紙, 再以丙酮及超音波清洗。
氧化動力學之測試矽將試片至於加熱爐中, 在大氣環境下, 定時取出紀錄恆溫氧化過程中之重量變化量。
3. 氧化試片的定性分析
A. XRD分析
氧化試驗後, 先以X光繞射儀分析氧化生成物, 分析時採用銅靶X光管, 其特性波長為1.542Å, 掃描速率0.05°/sec。
B. SEM-EDAX分析
試片在觀察之前先鍍上一層金膜, 使表面的氧化物能夠導電, 以利觀察氧化物的生長情形。
要分析試片橫截面, 必須先以無電鍍鎳(製作方法和溶液成份如後附錄所述)鍍上一層保護膜, 以避免在切割時氧化層剝落。
SEM觀察前, 試片應研磨至2000號砂紙, 在經1µm至0.05µm氧化鋁粉拋光, 在以超音波洗淨並浸蝕之後, 試片表面鍍上一層碳膜, 以便分析氧化層之型態以及氧化後元素重新分布的狀況。
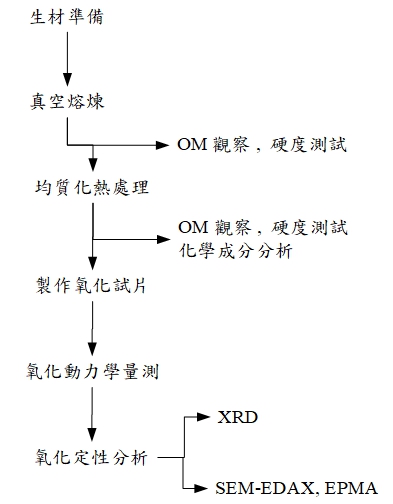
上述之實驗步驟, 可藉流程圖示意如下:
3. 實驗結果與討論
1. 顯微組織
A. 鑄造組織
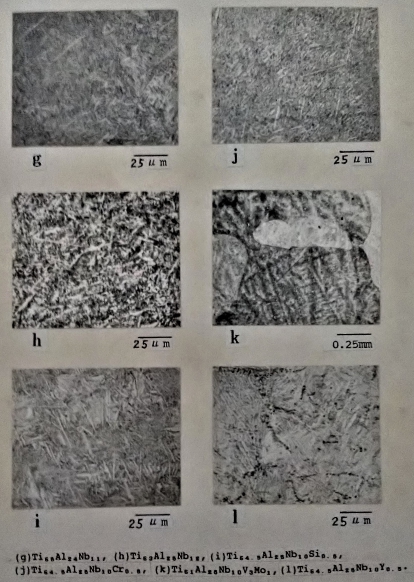
鈦鋁介金屬合金經過真空熔煉後之鑄造組織, 如圖16所示。在鋁含量不同的Ti-Al二元合金裡, 其組織有明顯的差異。Ti3A1合金從晶界延伸出雜亂的層狀組織, 參考Ti-Al的相圖(如圖17), 在1150℃附近有一包析(peritectoid)反應, 可能因此造成如此的鑄造組織。偏化合比的Ti₆₇Al₃₃合金相對地層狀組織則減少很多。Ti₅₇Al₄₃合金的金相是由細而雜亂的板狀(plate)組織所組成。而添加Nb的Ti3A1合金大都為羽毛狀的細化組織。

B. 均質化組織(如圖18所示)
Ti3A1和Ti₆₇Al₃₃合金先前的層狀鑄造組織, 經由均質化處理後已經消失, 取而代之的是粗大的晶粒。Ti₅₇Al₄₃合金的均質化組織, 有類似波來鐵(pearlite)的層狀結構, 根據相圖應該是α₂+γ兩相所組成的組織。
添加Nb的Ti3A1介金屬合金, 主要為籃網狀(basket-weave)的顯微組織; 並且含鈮量高的合金, 其組織也變得較細。


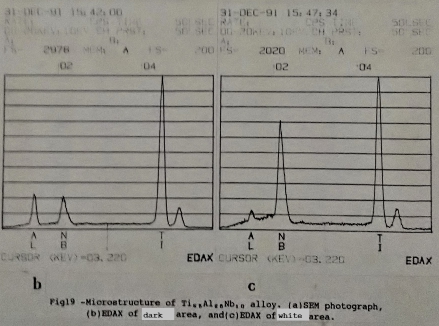
從掃描式電子顯微鏡(SEM)來觀察Ti3A1-Nb合金的組成分布。例如圖19的Ti₆₅Al₂₅Nb₁₀合金, 據EDAX的分析結果, 相片中的白色區域, 鈮含量比平均高出許多, 而鋁及鈦的量較少。灰色或暗色區域的鈮含量則比平均值略低。Kaufman [30]等人也發現β相的鈮含量比較高。同時, 鈮是使鈦鋁合金的高溫β相穩定的元素之一。因此可以確定經過1050℃的均質化處理之後, 所得到的組織是α₂相及殘留的β相所形成的。至於中間相如ω相及ο相則未加以證實。

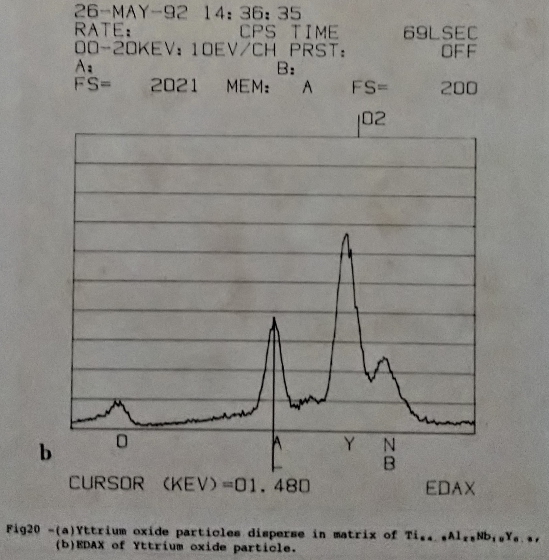
添加Si, Cr等第四元合金元素的Ti3A1-Nb合金與Ti3A1-Nb合金的組織相似。而添加Y的Ti3A1-Nb合金之鑄造組織在SEM下觀察結果顯示Y以氧化物顆粒的方式散布在基地(marix)中(如圖20)。
2. 鈦鋁介金屬合金之硬度測試及化學成份分析
鈦鋁介金屬合金的硬度值如表3所列。經過均質化處理後, 硬度均下降。而在Ti3A1合金的硬度壓痕周圍有不均勻的塑性變形出現。甚至Ti₆₇Al₃₃的成份合金, 其壓痕尖端附近嘿有裂紋產生(如圖21所示)。

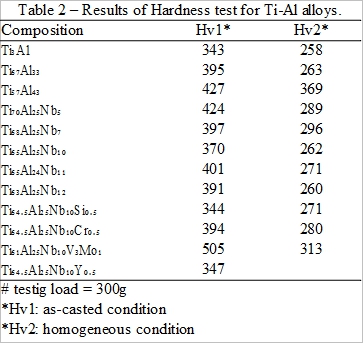
化學成份分析是由電子探針顯微分析儀(EPMA)坐定量分析及藉氧氮分析儀所測得的結果如附表4。合金成份均接近事先配製的組成, 同時含氧量介於800 ~ 1400ppm之間, 含氮量則在30ppm以下。另外, 在熔煉前後測其重量損失都小於0.08%。因此可以確定熔煉後的合金符合實驗所需的成份。
3. 氧化曲線
圖22顯示純Ti3A1介金屬合金在700℃的高溫氧化下, 經過150小時, 氧化層即有剝落的現象, 正如Lipsitt[1]所推測: 650℃是Ti3A1抗氧化溫度的上限。其餘兩種偏化合比的Ti3A1合金(Ti₆₇Al₃₃及Ti₅₇Al₄₃)因鋁含量較高而沒有剝落的痕跡, 如圖22所示。
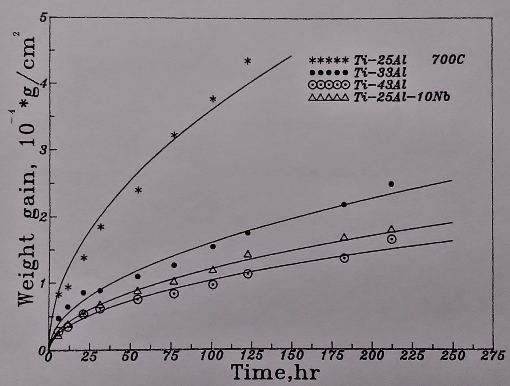
在圖22中亦顯示Ti₆₅Al₂₅Nb₁₀合金與前述二元鈦鋁介金屬合金比較, Ti3A1-Nb合金比Ti3A1和Ti₆₇Al₃₃兩合金之抗氧化性均較優異。所以, 添加Nb的合金效應顯示, 不僅可以改善合金的機械性質, 也增強了鈦鋁介金屬合金的抗氧化能力。Ti₆₅Al₂₅Nb₁₀較Ti₅₇Al₄₃的抗氧化能力略遜, 主要是因大量的鋁含量較Nb更為有效。
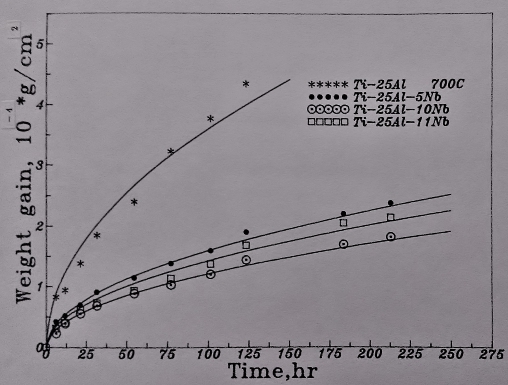
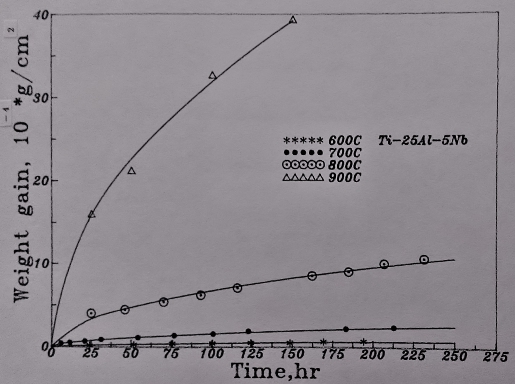
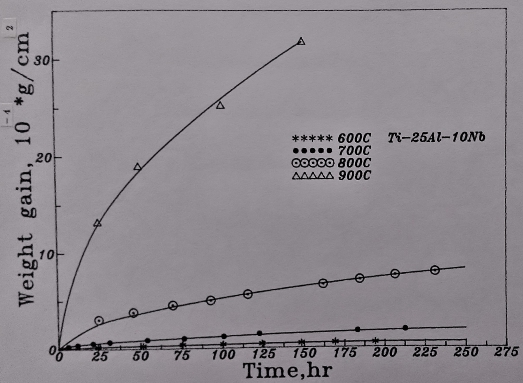
圖23表示Ti3A1-Nb介金屬合金在700℃的氧化曲線。結果顯示增加Nb的添加量對鈦鋁合金的抗氧化性甚有幫助。但Ti₆₅Al₂₅Nb₁₀合金比Ti₆₅Al₂₄Nb₁₁的氧化速率稍緩, 推測可能是Ti3A1-Nb介金屬合金中鋁的抗氧化貢獻比Nb來得更大所致。
第四元合金元素的添加對氧化速率的影響則如圖24所示, 添加Si和Cr有助於降低氧化速率; 但添加Y的效果卻有負面的影響, 液及加快了氧化的速度。已商業化的Super α₂合金(Ti₆₁Al₂₅Nb₁₀V₃Mo₁)也在同一溫度作比較, 發現其氧化速率最快, 暗示Ti3A1-Nb合金中添加V或Mo會不利於抗氧化能力。
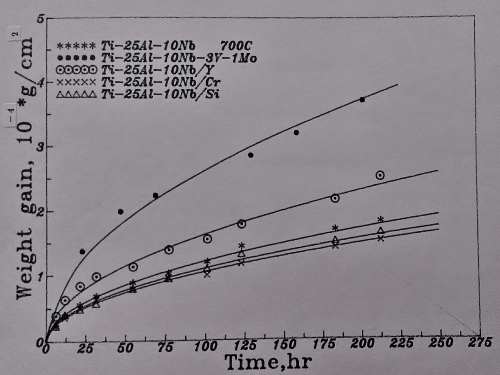
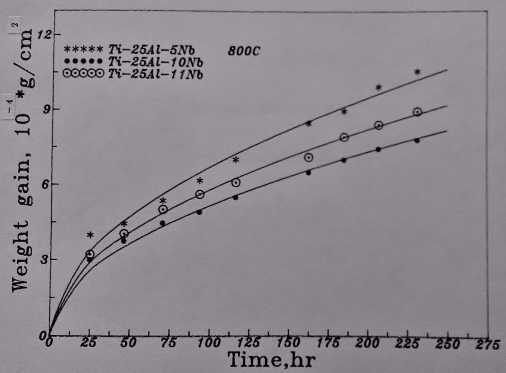
800℃時的氧化速率如圖25/26所示。同樣地, 抗氧化能力隨Nb含量的增加而提升。而且Ti₆₅Al₂₅Nb₁₀合金的抗氧化性仍較Ti₆₅Al₂₄Nb₁₁合金好。對於這種現象, 還未能提出一合理的解釋, 只能說合金中的鋁在此溫度範圍內(700~800℃)對氧化行為的影響比Nb更具優勢。
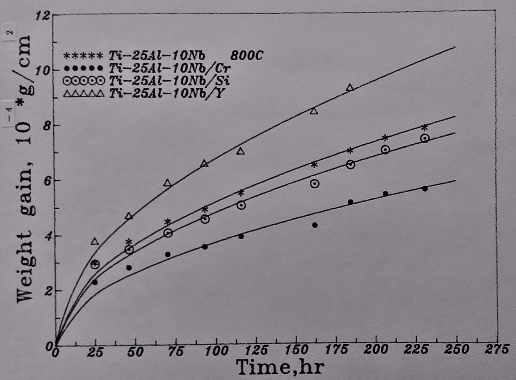
至於Si, Cr及Y的微量合金效應, 在800℃的高溫氧化的影響則更為明顯。添加Si和Cr的母材, 其氧化速率均減慢, 尤其是添加Cr的Ti3A1-Nb介金屬合金, 更能抑制氧化速率。而添加Y則氧化速度加快, 未能改善抗氧化能力。
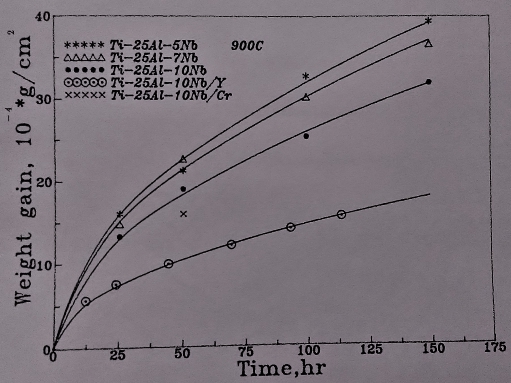
圖27表示的是Ti3A1-Nb介金屬合金的900℃的氧化曲線。與700℃/800℃的結果大致相同, 合金的氧化速率隨Nb的增加而降低。在第四元合金元素的影響方面, 添加Si和Cr者均不利氧化層的附著性, 量測氧化速率時, 超過50小時以上因發生氧化層剝落而無法紀錄。然而, 添加Y的Ti3A1-Nb合金在整個氧化過程中沒有發生氧化層剝落的狀況, 顯示其較佳的附著性, 同時氧化速率也因Y的添加而降低。此結果不同於700℃及800℃較低溫的氧化結果, 表示Y對Ti3A1-Nb介金屬合金的抗氧化性之影響需在較高溫才有幫助。
溫度對Ti3A1-Nb介金屬合金的高溫氧化之影響如圖28及29所示。600℃與700℃的氧化速率都很慢, 表示有不錯的抗氧化性。800℃以上氧化速率則急遽上升, 已喪失抗氧化的能力。
4. 表面氧化層的組織
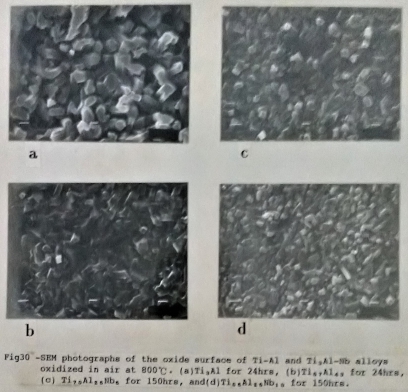
將Ti3A1經過800℃, 24小時的氧化後, 觀察其氧化物係呈現如柱狀晶(純為TiO₂)的生長型態,如圖30-(a)。而且氧化物晶粒之間留存較大的空隙, 有利於氧氣分子向材料內部擴散, 由此也可知Ti3A1合金的抗氧化性不佳。圖30-(b)是Ti₅₇Al₄₃合金在相同的氧化條件下的表面氧化物, 其氧化物分布較緻密。
圖30-(c)/(d)分別是經過800℃, 150小時氧化的Ti₇₀Al₂₅Nb₅及Ti₆₅Al₂₅Nb₁₀合金, 其表面氧化物的組織構造與圖30-(a)的Ti3A1合金比較, 顯示Nb有抑制氧化物成長的效果, 且因增加Nb的含量會使氧化物尺寸變小, 顯得更為緻密。
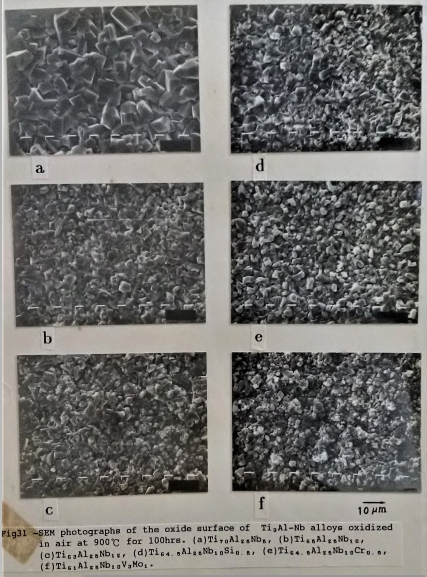
另外, 圖31/32分別是Ti3A1-Nb介金屬合金在900℃, 經過100小時及150小時的表面氧化物顯微組織。添加Si和Cr並沒有明顯改變Ti3A1-Nb合金的氧化物型態。而Super α₂合金的表面氧化物 顯得更為緻密。添加Y的Ti3A1-Nb合金則因為氧化釔成顆粒狀散佈於母材中, 而使表面氧化物有許多如花苞般的顆粒分布, 推測可能氧化釔顆粒提供氧化物有利的盛和及成長的位置, 才使表面的氧化物有如此不同的組織。
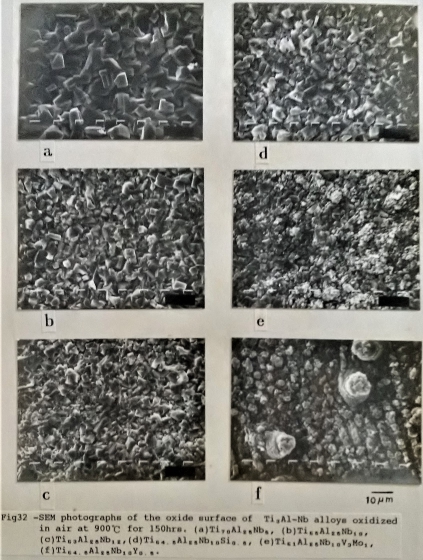
5. Ti3A1-Nb介金屬合金之氧化層組織
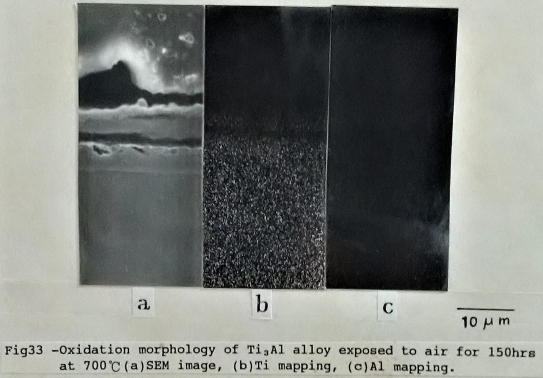
Ti3A1二元合金經過700℃氧化後, 氧化層約有10µm厚(如圖33), 最外層是無電鍍鎳所鍍上的鎳層, 其次是氧化層, 其大部分是氧化鈦(Rutile)及少許氧化鋁(α-A12O3)。並且氧化層中有凹陷的裂縫存在, 內部的氧化層之中也存在甚大的孔洞(void), 顯示Ti3A1合金在700℃以上已無法抵抗氧分子向內擴散進行氧化。
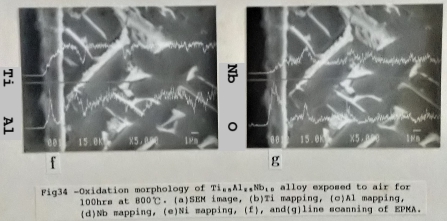
觀察Ti₆₅Al₂₅Nb₁₀合金, 在800℃及100小時的氧化層結構, 如圖34所示, 最外層是無電鍍鎳層, 然後是氧化鈦與大部分的氧化鋁混合的氧化層, 再來即是氧化鈦和少許的氧化鋁及氧化鈮的氧化層與金屬基地相鄰。估計此氧化層厚度約2-3µm厚, 推測是因形成較緻密的氧化層, 而使氧化速度減慢。另外, 從EPMA的線掃描顯示Ti3A1-Nb合金的β相中含氧量比α₂相要高, 表示β相容易吸氧, 若與氧化層相接, 易造成內部氧化(internal oxidation)。
Ti₇₀Al₂₅Nb₅合金經過900℃, 100小時的氧化, 最外層的氧化層幾乎全部為疏鬆的氧化鈦所組成, 緊接著的是氧化鋁層, 在其次為氧化鈦和氧化鈮相混的氧化層, 而氧化層與金屬基地之間有一層極薄的富鋁及鈮的氧化層。如圖35/36所示。而添加10at% Nb的Ti3A1合金, 在相同的氧化條件下, 其氧化層的分布(如圖37), 由外向內依序為: 氧化鋁和氧化鈦的混合層, 其次是氧化鈦與氧化鈮的氧化層, 然後是一層薄薄的氧化鋁層和金屬基地相接。
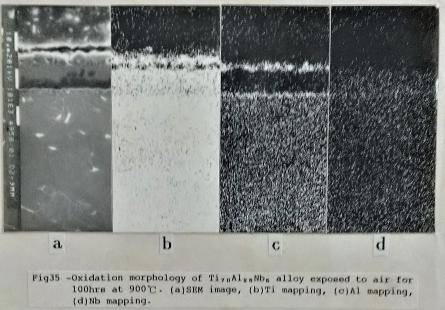
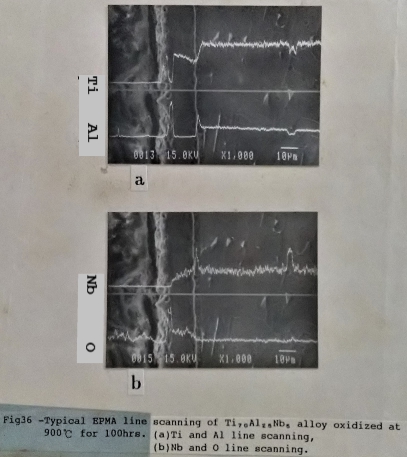
根據以上的描述, Nb的加入Ti₃A1介金屬合金中能促使 A12O3氧化層的形成, 能有效地阻止氧分子相材料內部擴散。比較Ti₆₅Al₂₅Nb₁₀合金在800℃形成的氧化層與Ti3A1合金在700℃形成的氧化層即可得知。再進一步探討Nb添加量的多寡對氧化層結構的影響; 可比較Ti₆₅Al₂₅Nb₁₀合金與Ti₇₀Al₂₅Nb₅合金(如圖35/37所示)的氧化層, Ti₇₀Al₂₅Nb₅合金最外層是疏鬆的TiO₂氧化層, 沒有保護的作用。在TiO₂氧化層底下是一層薄的A12O3氧化層, 從EPMA的線掃描發現氧分子被阻礙在此層, 只有少量擴散進入內部。但Ti₆₅Al₂₅Nb₁₀合金的最外氧化層已有氧化鋁形成, 而不像Ti₇₀Al₂₅Nb₅合金外層疏鬆的氧化鈦。顯示增加Nb含量不僅促進A12O3的生成, 同時使鋁原子擴散加快, 在最外層形成較緻密的A12O3+ TiO₂氧化層, 有較佳的抗氧化性。
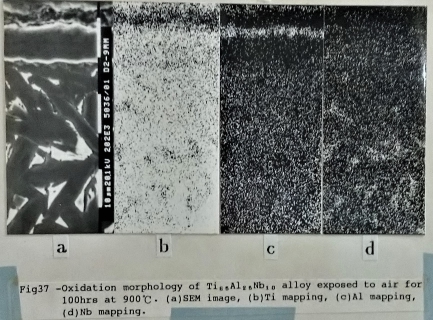
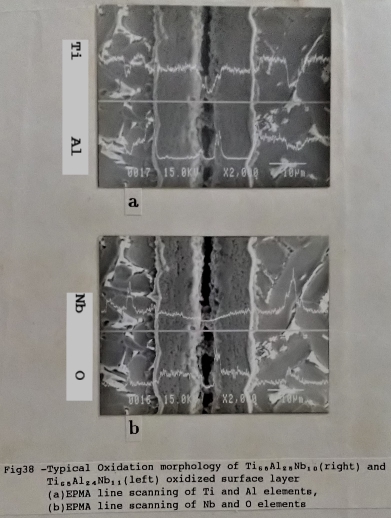
先前討論Ti₆₅Al₂₅Nb₁₀與Ti₆₅Al₂₄Nb₁₁兩者氧化速率的差異時, 曾顯示Ti₆₅Al₂₅Nb₁₀合金的抗氧化性比Ti₆₅Al₂₄Nb₁₁要好, 從橫截面氧化層的掃描來觀察, 如圖38所示, 右側為Ti₆₅Al₂₅Nb₁₀, 左側為Ti₆₅Al₂₄Nb₁₁合金, 由鋁元素及氧元素的分布情形比較, Ti₆₅Al₂₅Nb₁₀合金的最外層的A12O3較Ti₆₅Al₂₄Nb₁₁合金來得多。而在Ti₆₅Al₂₄Nb₁₁合金的氧化層內, 並無如Ti₆₅Al₂₅Nb₁₀合金之氧化層中氧元素被阻擋而堆積的現象出現(形成A12O3薄膜, 但無連續性), 這也是可能是兩合金在抗氧化能力上有所差別的原因。(以上EPMA掃描之差異經過不同位置測試確定均有類似的現象出現)
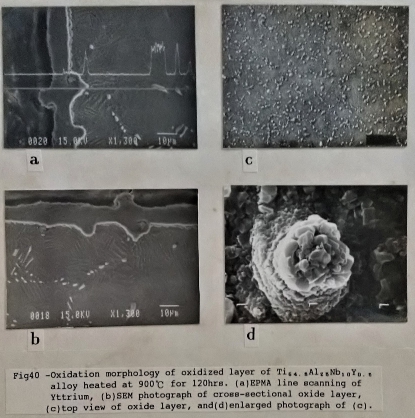
在添加第四元合金元素的Ti3A1-Nb介金屬合金的氧化層結構的研究中, 添加Si與Cr者, 由於添加量甚微, 只能從氧化速率看出對合金的影響。在氧化層組織上與Ti₆₅Al₂₅Nb₁₀合金無多大差別。但是添加Y的Ti3A1-Nb合金則因為Y是以氧化物顆粒散佈於母材中, 因此形成較奇特的氧化層結構。在SEM下觀察, 對照其表面氧化層與截面的氧化層(如圖39/40), 表面的花苞狀氧化物向外突出, 又因氧化釔顆粒會促使氧化層相內成長, 在金屬與氧化層之間形成凹凸不平的平面,
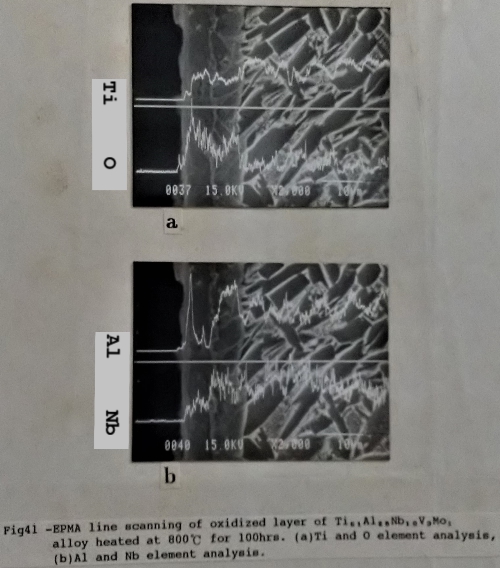
可能也因此加強了氧化層的附著力, 亦即第二章所提到的釘掛效應(Pegging or Keying effect)。Super α₂的氧化層也比較特別, 除了Ti3A1-Nb合金的氧化層外, 在靠近氧化層的母材形成一內部氧化層(如圖41), 同時鋁和鈮的濃度在內部氧化層比較高。顯然Super α₂形成氧化層的方式與其他Ti3A1-Nb合金有別, 但氧化機構為何? 尚有待進一步的研究。

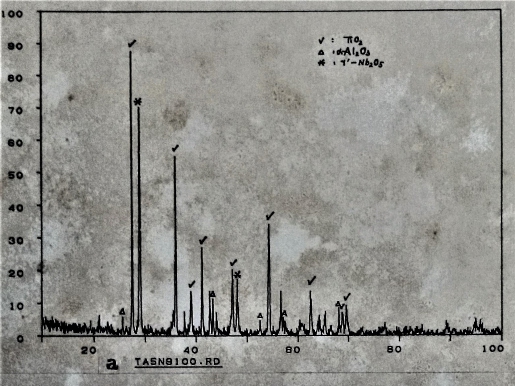
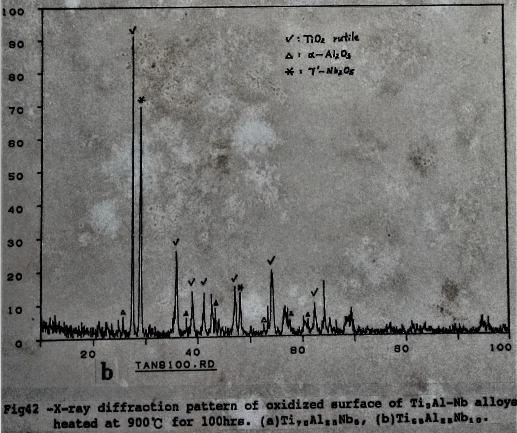
藉X光繞射亦可確定氧化物的組成。圖42是Ti₆₅Al₂₅Nb₁₀的表面氧化物以X光繞射分析, 分析結果得知由TiO₂(Rutile), α-A12O3, γ-Nb₂O₅三種化合物組成氧化層。
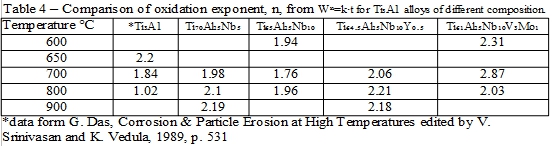
6. 氧化指數, n
第二章第二節曾提到氧化速率方程式, Wⁿ=k∙t。式中的氧化指數, n, 若有變化則分別代表不同的氧化機構, 所以可由n值來判斷為何種氧化膜式。只要將 Wⁿ=k∙t方程式兩邊取對數, 以ln(W) vs. ln(t)作圖計算斜率即可得出實際的氧化指數, 將實驗數據依照此法求得n值列於表4, n值約為2左右, 可知Ti3A1-Nb合金在實驗的溫度範圍大致符合以離子擴散為控制機構的拋物線型氧化方式, 即n=2。
7. Arrhenius關係圖與氧化活化能
由於Ti3A1-Nb介金屬合金是大致遵守拋物線型的氧化方式, 因此可由第一節中氧化速率的曲線求出kₚ氧化常數, 再配合Arrhenius關係式:
kₚ=k0∙exp(-Q/RT) ..........(9)
取對數, log(kₚ) 對溫度倒數(1/T)作圖, 即可得Arrhenius圖, 如圖43/44, 圖中右上及左下的直線各是氧化鈦及氧化鋁的形成動力學直線。Ti3A1合金的直線是較靠近氧化鈦的一邊, 而添加Nb之後, Ti3A1-Nb合金則向形成氧化鋁的方向靠近, 證明Nb的合金效應可以促使 A12O3生成, 亦可由前述的氧化層結構對照得知。
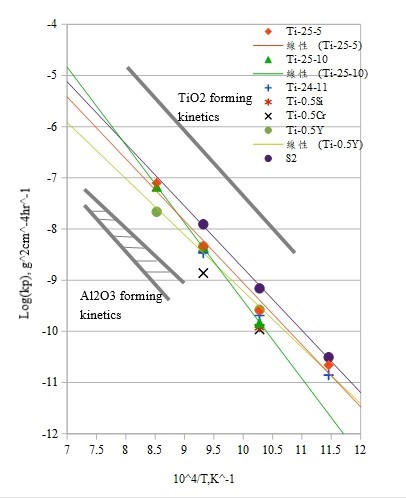
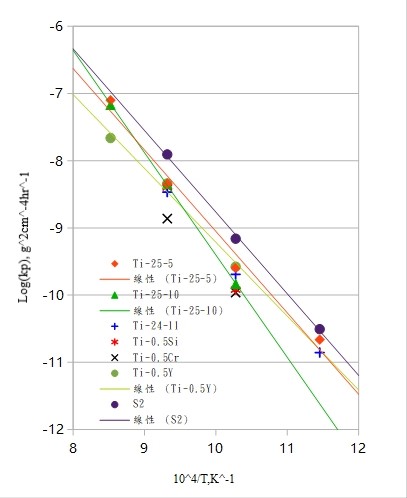
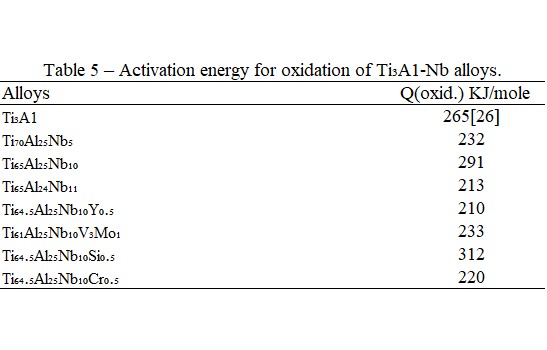
在Arrhenius圖中求其斜率即可得氧化所需之活化能, Qoxid., 列於表5, 添加Nb的Ti3A1合金, 其氧化活化能與Ti3A1合金比較, 隨Nb量的增加呈現先降後升的趨勢。類似鈦鋁合金的氧化活化能的變化, 在增加鋁含量時會使氧化加速, 降低氧化所需的活化能。G.Welsch認為這種現象是dopping的效應, 但添加Nb的Ti3A1合金是否如此, 則有待商榷。
5. 結論
1. Ti3A1介金屬合金因添加Nb而使氧化速率降低, 並且抗氧化性隨Nb添加量的提高而增加。然而鋁的抗氧化能力較鈮更好, 依結果顯示, 鋁含量的多1%可使Ti₆₅Al₂₅Nb₁₀合金的抗氧化能力比Ti₆₅Al₂₄Nb₁₁合金更佳。
2. Si及Cr等第四元合金元素的添加有助於Ti3A1-Nb介金屬合金抗氧化能力的增進。添加Y可以改善氧化層之附著性; 然而Y對氧化速率的降低則在較高溫度時才有作用。
3. 從氧化層的結構來看, Nb的添加可以細化表面氧化物的晶粒, 形成較緻密的氧化層, 且可加速鋁元素的向外擴散, 促進A12O3氧化層的生成。
4. Ti3A1-Nb介金屬合金的氧化機構係以離子擴散控制為主, 亦即n=2之拋物線型氧化方式。
附 錄:
無電鍍鎳之實驗步驟, 條件及配方如下所列:
1. 粗化: 2 wt% HF 時間: 15 min
40 g/L NH₄F
0.1 g/L SnCl₂ 溫度: 60℃
40 g/L KCl
2. 活化: 20 g/L SnCl₂ 時間: 7-9 min
40 cc/L HCl
3. 敏化: 0.25 g/L PdCl₂ 時間: 10 min
2.5 cc/L HCl
4. 無電鍍: 28.09 g/L NiSO₄∙7H₂O
21.2 g/L NaH₂PO₂∙H₂O
27.01 g/L Na₂C₄H₄O₄∙6H₂O
pH=5, 溫度: 88℃
註: 依個人經驗, Ti3A1-Nb合金只需後面的三個步驟即可。
參考文獻
[1] H.A.Lipsitt, High Temperature Ordered Intermetallic Alloys, C.C.Koch, C.T.Liu, and N.S.Stoloff, eds., MRS Pittsburgh, PA 1985, p. 351.
[2] H.A.Lipsitt et al., Metall. Trans., A, vol. 11A, 1980, p.1369.
[3] Y.Umakoshi et al., J. Mater. Sci., vol. 24, 1989, p. 1599.
[4] Y.Shida and H.Anada, J. Japan Inst. Metals, vol.55, No.6, 1991, p. 690.
[5] 吉原美知子, 田中良平: 日本金屬學會會報, 30(1991), p. 61.
[6] E.Kobayashi et al., High Temp. Tech., vol. 8, No. 3, August, 1990, p. 179.
[7] K.Kasahara et al., J. Japan Inst. Metals, vol.54, No.8, 1990, p. 948.
[8] S.M.L.Sastry and H.A.Lipsitt, Ti 80, Science and Technology, H.Kimura and O.Izumi, eds., TMS-AIME, Warrendale, PA, 1980, p. 1231.
[9] S.M.L.Sastry and H.A.Lipsitt, Metall. Trans., vol. 8A, 1977, p. 1543.
[10] 洪衛朋, 博士論文, 國立臺灣大學材料科學及工程研究所, 1991.
[11] D.Banerjee et al., Acta Metall., 36, No. 4, 1988, p. 871.
[12] R.Strychor et al., Metall. Trans., vol. 19A, 1988, p. 225.
[13] A.K.Gogia et al., Metall. Trans., vol. 21A, 1990, p. 609.
[14] K.Muraleedharam et al., Metall. Trans., vol. 23A, 1992, p. 401.
[15] M.Khobaib et al., 2nd international SAMPE Metals confrence, Aug. 2-4, 1988, p. 262.
[16] J.Subrahmanyam, J. Mater. Sci., vol. 23, 1988, p. 1906.
[17] H.A.Lipsitt, Nicholas J. Grant symposium, June 16-18, 1985, p. 157.
[18] 吳易座, 碩士論文, 國立臺灣大學材料科學及工程研究所, 未發表.
[20] G.H.Meier and N.Birk, “Introduction to High Temperature Oxidation of Metals”, Edward Arnold, London, 1983.
[21] Wagner, C., Z. Phys. Chem. Vol. 21, 1933, p. 25.
[22] K.L.Luthra, Oxidation of Metals, vol. 36, Nos 5/6, 1991, p. 475.
[23] G.Welsch & A.I.Kahveci, “Oxidation of High-Temperature Intermetallics” edited by T.Grobstein & J.Doychak, the Minerals, Metals & Materials Society, 1989, p. 207.
[24] 笠原和男, 橋本健紀, 土肥春夫, 過本得藏: 日本金屬學會會誌, 53(1989), p. 58.
[25] G.Das and F.W.Vahldiek, “Corrosion & Particle Erosion at High Temperature” edited by V.Srinivasan and K.Vedula, the Minerals, Metals & Materials Society, 1989, p. 531.
[26] J.Stringer and P.Y.Ho, ibid, p. 383.
[27] T.A.Ramanarayanan et al., Oxidation of Metals, vol. 29, Nos 5/6, 1988, p. 445.
[28] D.P.Whittle and J.Stringr, Phil. Trans. R. Soc. Lon. A295, 1980, p. 305.
[29] 洪衛朋, 碩士論文, 國立臺灣大學材料科學及工程研究所, 1987.
[30] M.J.Kaufman et al., Scripta Metall. Vol. 23, 1989, p. 1697.
誌 謝
這一年的研究歲月裡, 雖然遇到一些困難與挫折, 但是還是經過自我不斷的嚐試及許多人的幫助, 才得以順利完成實驗。因此在這裡必須感謝技術員盧錫全和高崇源, 在試片分析上給予許多的協助; 以及同學李定年, 蕭志祥等人在精神上和實驗上的鼓舞和幫助。最後, 更感謝指導教授 顧鈞豪博士兩年來課業的教誨以及論文的指正, 使得在臺大的研究生涯不致虛渡。
謹以此成果獻給我親愛的家人。


