當你買食品、營養保充品,甚至化妝品,產品有時會標示GMP及cGMP的認證。
GMP 及 cGMP是針對藥廠所定下的規範,跟一般廠房是不一樣的:GMP 及 cGMP是對產品質素的一種保證:作為消費者,認識這些廠房規範對選購產品有絕對的幫助! https://blog.udn.com/alpineatks/178424466
文章重點:
- GMP 是甚麼?
- cGMP 是甚麼?
- GMP v.s cGMP(5大差別)
- 有GMP,cGMP的食品、藥品及代妝品的例子
- 其他常見認證
GMP是甚麼?

GMP 全名 Good Manufacturing Practice ,中文良好作業規範,是針對藥品、醫療產品生產及質量管制的準則。
有些食品或保健食品廠為了提高消費者信心,也會升級做GMP廠房標準,但不是必須的。
背景

1969年,由世界衛生組織WHO開如始製定GMP,列明全球食品及藥品的製作規範。
目的
- 確保產品的質素一致性
- 控制藥品以乎合其預其用途的質量標準,並按照產品規格
- 定義,驗證,審查和紀錄及測試產品製作過程
- 確保廠房人員、場所、設備及材料適合藥品、醫療製品包括疫苗的生產
- 其法律效力以約束不合乎要求的藥廠
好處
- 減少藥品製造過程的風險,包括交叉污染及容器貼上錯誤標籤
- 把造藥過程規範化及國際化,增加消費者對藥品的信心
不同地區的GMP
現今全球超過100個國家在WHO的GMP列出的條件下,製作屬於自己國家的GMP準則。
cGMP 是甚麼?

cGMP全名Current Good Manufacturing Practice,中文現行藥品生產管理規範,表示GMP藥廠的生產作業過程是與最新標準同步的狀態,是要求更嚴格的作業規範。
有些食品或保健食品廠為了提高消費者信心,也會升級做cGMP廠房標準,但不是必須的。
背景

由美國FDA於1998件草擬的藥品製作規範;現於美國,歐洲及日本都用cGMP的規範。
目的
- cGMP中的c代表現行,即是要求藥廠的技術及系统是最新並合乎標準!
- 確保穩定及安全的產品質量!
- 確保廠房的設施,原材料,製作及加工過程,包裝方法合乎cGMP的最低標準!
- 嚴格監管整個管理系统,確保工作人員跟上cGMP的規範
- 確保藥品乎合其聲稱的藥品功效!
好處
- 對藥品的質素要求更嚴格,令消費者更有信心!
cGMP v.s GMP
驟看之下,cGMP及GMP分別不大,當你仔細看看WHO及FDA定下的準則,兩者對硬件及軟件的監管標準其實並不一樣!
表一 :cGMP 及 GMP廠的5大不同

cGMP對硬件,即是廠房設備,設施,藥品生產,場地衛生、包裝及標簽等等都要最新並合乎規格的!
廠房場地的通風系统,衛生情況,設施的保養對藥品質素影響甚大,因此cGMP對其有嚴格的要求!
某些地區如中國大陸採用的GMP準則,在硬件方面跟cGMP是差不多的,因為地區可以在WHO GMP的框架下定立自己的作業規範。
反而兩者最大分別是對軟件方面,即是生產過程,工作人員的職責規定。
因為人為的錯誤較難控制,所以FDA的cGMP對生產過程及人員職責有更嚴格細緻的規定,以確保產品的質素及生產過程的精準度。
有GMP, cGMP認證的食品,保健品及代妝品的例子!
藥品及醫療產品對人體健康有很大風險,因此必須有GMP或cGMP的認證。
那麼一般食品、保健食品、營養補充品及代妝品呢?是不是有GMP或cGMP認證會比較好?
答案當然是。
而升格做GMP或更嚴格的cGMP廠房需要的成本更高,要經過審查及定期審查,所以不是每一日食品廠都願意做。
有GMP認證品牌
藥品或保健品:余仁生、位元堂、維特健靈等等
化妝及護膚品:Shiseido、彩豐行等等


有cGMP認證品牌
藥品或保健品:森下仁丹、澳美製藥、Healthy care等等

其他常見食品及藥品認證
食品包括一般包裝食品,健康食品及營養保充品;藥品就是有醫療用途的藥物;
除了定義廠房規格的GMP, cGMP之外,還有一些確認食品及藥品能否符合標準的認證。
認證是什麼?
認證是由第三方對產品及服務,過程,系统及人員發出證書,以證明產品符合特定要求:認證主要分兩種:產品認證和系统認證! https://blog.udn.com/alpineatks/178424466
產品認證
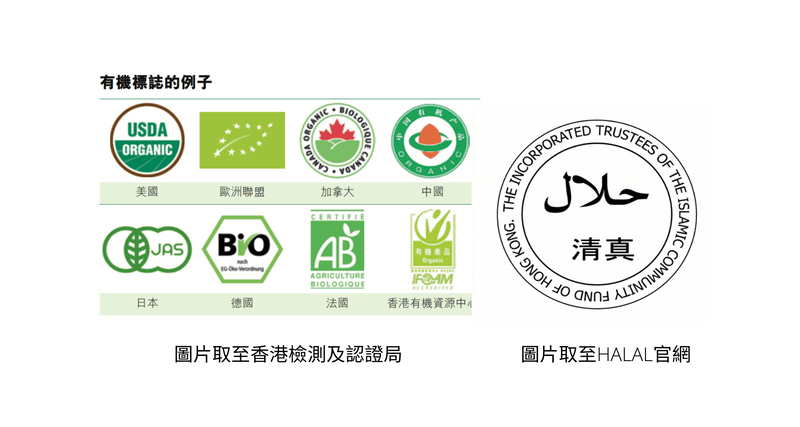
針對產品特性的認證
- 有機食品認證:例如美國的USDA Organic;
- 清真食品認證:
- 猶太食品認證:等等
產品認證
。 https://blog.udn.com/alpineatks/178424466
系统認證

針對機構管理的不同範疇:
- 食物安全管理 ISO22000
- 品質管理 ISO 9001
- 環境管理 ISO 14001
- 能源管理 ISO 50001
- 食品安全體系認證FSSC22000
- 危害分析安全管理體系HACCP
等等,等等系统認證! https://blog.udn.com/alpineatks/178424466
*資料來源:
世界衛生組織 WHO、美國食物及藥物管理局FDA、香港檢測及認證局、品牌官方網站! https://blog.udn.com/alpineatks/178424466